NWChem: Parameter Sweep and Pre/Post-processing
Even if you are not interested in Chemistry, this tutorial illustrates several important Balsam concepts:
- Setting up an Application that requires several modules to run
- Pre-processing to generate input files
- Post-processing to parse and store calculation results
- Storing JSON data with PostgreSQL
- Creating a large parameter sweep-type ensemble
- Adding a reduce-type (summary) job using
dag.add_dependency()
Water HF/6-31G Potential Energy Scan
In this exercise, we'll use NWChem on Theta to calculate the electronic ground state energy of the water molecule. We would like to repeat this calculation many times as a function of the symmetric O--H bond stretching distance, in order to generate a 1-dimensional potential energy surface (PES).
Let's see how an existing build of NWChem on Theta (courtesy of Alvaro Vazquez-Mayagoitia) can be used in a Balsam workflow to generate the water PES.
Setting Up
Let's create a clean workspace and Balsam DB for this exercise as follows.
$ rm -r ~/.balsam # reset default settings (for now)
$ mkdir ~/tut-nwchem
$ cd ~/tut-nwchem
$ module load balsam
$ balsam init db
$ . balsamactivate db
The NWChem Application
Suppose we were looking for a public build of NWChem on Theta. We search in
/soft/applications and find the binary
/soft/applications/nwchem/6.8/bin/nwchem alongside a submit.sh script to
launch a run. In order for NWChem to run properly, this script loads some
modules and sets many MPICH environment variables.
The easiest way to create this NWChem environment is Balsam is to copy all of
the module load and export statements into the envscript of our NWChem
ApplicationDefinition. We first copy over the file: cp
/soft/applications/nwchem/6.8/bin/submit.sh envs.sh and then delete the
launch commands, so it's only setting up the environment like this:
#!/bin/bash
# envs.sh
module add atp
module add intel
export MPICH_GNI_MAX_EAGER_MSG_SIZE=16384
export MPICH_GNI_MAX_VSHORT_MSG_SIZE=10000
export MPICH_GNI_MAX_EAGER_MSG_SIZE=131072
export MPICH_GNI_NUM_BUFS=300
export MPICH_GNI_NDREG_MAXSIZE=16777216
export MPICH_GNI_MBOX_PLACEMENT=nic
export MPICH_GNI_LMT_PATH=disabled
export COMEX_MAX_NB_OUTSTANDING=6
Instead of defining applications from the command line, we can write a small Python script:
#!/usr/bin/env python
# apps.py
'''Register the two Applications'''
import os
from balsam.core.models import ApplicationDefinition as App
if not App.objects.filter(name="nwchem-water").exists():
nwchem = App(
name = 'nwchem-water',
executable = '/soft/applications/nwchem/6.8/bin/nwchem',
preprocess = os.path.abspath('pre.py'),
postprocess = os.path.abspath('post.py'),
envscript = os.path.abspath('envs.sh'),
)
nwchem.save()
if not App.objects.filter(name="plot-pes").exists():
plotter = App(
name = 'plot-pes',
executable = os.path.abspath('plot.py')
)
plotter.save()
You'll notice this script provides pre- and post-processing scripts for nwchem-water, as well as a second plot-pes app. We will get to these shortly. First, let's write this plot-pes script.
The "Plotting" Application
After our PES scan is completed, we would like to generate a summary plot of the data. This can be accomplished by adding a plot-pes task as the child of all the nwchem-water tasks. Creating these dependencies will form a DAG, as shown in the figure below. The plot step only runs after all the parent NWChem calculations have finished succesfully.
The DAG consists of several independent NWChem single-point energy tasks, followed by a data-summarization task that depends on successful completion of all the energy calculations.
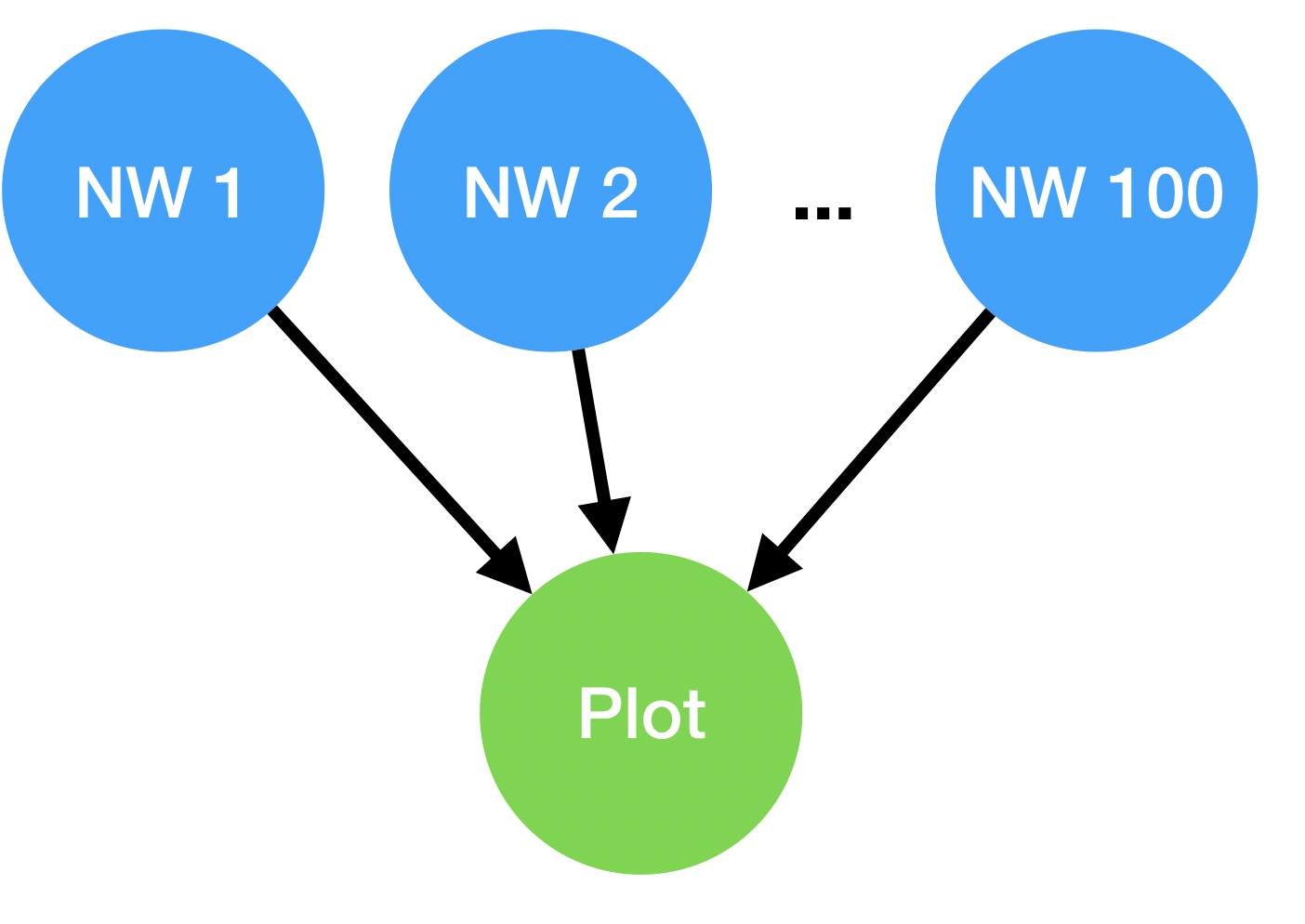
Now create the plot script as shown below. The highlighted lines show how the task will find its parent tasks through the Python API. The results are organized by pulling O--H bond length (r) and the electronic energy (energy) from each nwchem-water task's data dictionary.
#!/usr/bin/env python
# plot.py
'''Summarize results in plottable text format'''
from balsam.launcher.dag import current_job
results = [
(task.data['r'], task.data['energy'])
for task in current_job.get_parents()
]
results = sorted(results, key=lambda pair: pair[0])
print("%18s %18s" % ("O-H length / Ang", "Energy / a.u."))
for r,e in results:
print("%18.3f %18.6f" % (r,e))
Creating the Workflow
We can write a simple script to create one instance of this workflow. The steps include:
- Defining a grid of r values
- Creating a nwchem-water task for each r value
- Creating a plot-pes task
- Adding a Dependency from each nwchem-water parent task to the plot-pes task
The script should look as follows. One line is highlighted for each of the four steps listed above. You will notice that we are setting the data attribute in the BalsamJob constructor for each nwchem-water task. This field can hold arbitrary JSON data; it is stored efficiently in a Postgres binary format (JSONB) and is readily queried with Django.
#!/usr/bin/env python
# populate.py
'''Add Water potential energy scan tasks to DB'''
from balsam.launcher.dag import BalsamJob, add_dependency
import numpy as np
r_grid = np.linspace(0.8, 1.3)
water_scan = []
for i, r in enumerate(r_grid):
job = BalsamJob(
name=f"task{i}",
workflow="demo",
description=f"r = {r:.3f}",
application = "nwchem-water",
args = "input.nw",
num_nodes = 1,
ranks_per_node = 64,
cpu_affinity = "depth",
data = {'r':r, 'theta': 104.5},
)
water_scan.append(job)
job.save()
plotjob = BalsamJob(
name="plot",
application="plot-pes",
workflow="demo",
input_files="",
)
plotjob.save()
for job in water_scan:
add_dependency(parent=job, child=plotjob)
Preprocess: Generating NWchem input
We need to create an input file for each NWChem calculation before it
runs. We can do this in the preprocess script
pre.py that is used in the nwchem-water ApplicationDefinition. This script:
- uses
balsam.launcher.dag.current_job{.interpreted-text role="bash"} to grab the context of the current job - reads the internal coordinates (r, theta) from the task data field
- converts to Cartesian (xyz) coordinates
- writes-out an input deck for a Hartree Fock calculation in the 6-31G basis
The four steps above are mapped to the highlighted lines in the script below:
#!/usr/bin/env python
# pre.py
'''Generate input file for NWChem'''
import numpy as np
from balsam.launcher.dag import current_job
def water_cartesian(r, theta):
cost = np.cos(np.radians(theta))
sint = np.sin(np.radians(theta))
O = ['O', 0.0, 0.0, 0.0]
H1 = ['H', r, 0.0, 0.0]
H2 = ['H', r*cost, r*sint, 0.0]
return (O,H1,H2)
def input_deck(cartesian_coords):
coords = [' '.join(map(str, c)) for c in cartesian_coords]
return f'''
start h2o
title "Water in 6-31g basis"
geometry
{coords[0]}
{coords[1]}
{coords[2]}
end
basis
* library 6-31g
end
task scf
'''
data = current_job.data
r, theta = data['r'], data['theta']
coords = water_cartesian(r, theta)
with open('input.nw', 'w') as fp:
fp.write(input_deck(coords))
Note
Pre- and Post-processing scripts run in the task's working directory, so we don't have to worry about absolute paths when opening files.
Postprocess: Parse and store NWChem output
The last piece of our workflow is the post.py script responsible for
collecting NWChem outputs after each nwchem-water task finishes.
Note that that the output (both stdout and stderr) of each task are directed
to a file named {job.name}.out.
We simply scan through the lines of this file and extract the final HF
energy. Finally, we store it in the JSON data field so the plot-pes
step can find this result at the end.
#!/usr/bin/env python
# post.py
'''Parse Energy from NWChem output'''
from balsam.launcher.dag import current_job
outfile = current_job.name + ".out"
energy = None
with open(outfile) as fp:
for line in fp:
if 'Total SCF energy' in line:
energy = float(line.split()[-1])
break
current_job.data['energy'] = energy
current_job.save()
Note
Post-processing scripts can also be used to programatically handle
errors. This is enabled by setting the post_error_handler=True flag in
the BalsamJob, and inspecting the current_job.state in the
postprocessor. In this tutorial, the postprocess is only invoked after
successful completion of runs.
Putting it All Together
Now we can populate the DB with our ApplicationDefinitions and workflow,
then submit a job and watch it go. We need to be sure that our scripts
are executable, since our scripts are registered directly as executables.
It would also have been fine to set python /path/to/pre.py as the preprocess,
without making pre.py itself executable, for instance.
$ chmod +x *.py # set exe permission
$ python apps.py # populate apps
$ balsam ls apps --verbose # check apps in DB
$ python populate.py
$ balsam ls # check jobs in DB
$ balsam submit-launch -n 5 -t 60 -A Project -q Queue --job-mode=mpi
When the workflow starts running, the Balsam command line is a great way to quickly navigate the tasks and see the status of everything in realtime. Follow along to learn some of the navigation tricks below:
# count up jobs by state
$ balsam ls --by-states
JOB_FINISHED 10
RUNNING 5
PREPROCESSED 85
AWAITING_PARENTS 1
# list finished jobs only
$ balsam ls --state JOB_FINISHED
job_id | name | workflow | application | state
--------------------------------------------------------------------------------------
159070cb-03f8-4a37-9ed1-66bfb906e4bb | task4 | demo | nwchem-water | JOB_FINISHED
db12a2e7-f8b1-4eb5-9c03-5bb0c4ac74f8 | task3 | demo | nwchem-water | JOB_FINISHED
72d7f99e-76a5-495d-9d5f-648e0582e3a4 | task2 | demo | nwchem-water | JOB_FINISHED
# change directory (cd) to task with job_id starting with 1590
$ . bcd 1590
$ ls
h2o.movecs h2o.p h2o.zmat input.nw postprocess.log preprocess.log task4.out
# This one is cool: use BALSAM_LS_FIELDS to add "data" to the table display
# Then list finished jobs to see the results at a glance!
$ BALSAM_LS_FIELDS=data balsam ls --state JOB_FINISHED
job_id | name | workflow | application | state | data
--------------------------------------------------------------------------------------------------------------------------------------------------------------
159070cb-03f8-4a37-9ed1-66bfb906e4bb | task4 | demo | nwchem-water | JOB_FINISHED | {'r': 0.8408163265306123, 'theta': 104.5, 'energy': -75.949402426371}
db12a2e7-f8b1-4eb5-9c03-5bb0c4ac74f8 | task3 | demo | nwchem-water | JOB_FINISHED | {'r': 0.8306122448979593, 'theta': 104.5, 'energy': -75.941790242862}
72d7f99e-76a5-495d-9d5f-648e0582e3a4 | task2 | demo | nwchem-water | JOB_FINISHED | {'r': 0.8204081632653062, 'theta': 104.5, 'energy': -75.93319131096}
e667e9e8-b77d-4e78-9b73-f5084cc62e3a | task7 | demo | nwchem-water | JOB_FINISHED | {'r': 0.8714285714285714, 'theta': 104.5, 'energy': -75.966950538242}
77ecaf92-e462-49b2-8823-2d5a9c449528 | task49 | demo | nwchem-water | JOB_FINISHED | {'r': 1.3, 'theta': 104.5, 'energy': -75.863011732378}
6ca50987-d509-43aa-9604-da1d0d625a82 | task3 | demo | nwchem-water | JOB_FINISHED | {'r': 0.8306122448979593, 'theta': 104.5, 'energy': -75.941790242862}
46f689e1-7a1a-49b7-bc74-4d058b6faa1b | task19 | demo | nwchem-water | JOB_FINISHED | {'r': 0.9938775510204082, 'theta': 104.5, 'energy': -75.981078202929}
b83a6cb2-5ad9-44aa-b28b-d665bbbfbedf | task10 | demo | nwchem-water | JOB_FINISHED | {'r': 0.9020408163265307, 'theta': 104.5, 'energy': -75.977730477137}
b618dcbf-1e49-46e9-9b2f-b0ee69904dfc | task1 | demo | nwchem-water | JOB_FINISHED | {'r': 0.8102040816326531, 'theta': 104.5, 'energy': -75.923535853437}
ac765c84-6544-4bf0-9f60-b898cb6ec117 | task2 | demo | nwchem-water | JOB_FINISHED | {'r': 0.8204081632653062, 'theta': 104.5, 'energy': -75.93319131096}
# Check on the Plot step
$ balsam ls --name plot
job_id | name | workflow | application | state
------------------------------------------------------------------------------
fce9ecb3-b025-4e18-b499-a5c21e97e4ee | plot | demo | plot-pes | JOB_FINISHED
# Change to the plot dir
$ . bcd fce9
$ cat plot.out