Containerizing Single Node MPI Applications for High Throughput
In this article, I'll demonstrate how MPI applications can be
containerized and run under the Balsam serial job mode on
Theta. This approach enables significantly higher throughput for a
large number of single-node jobs that use MPI to scale across the cores
of each KNL node. This is particuarly important for users of certain
applications like NWChem, which were designed to scale to many nodes in
a distributed-memory paradigm. If you are nevertheless interested in
doing high-throughput computing with these applications, for instance to
generate a large dataset for machine learning, then containerizing them
with a generic MPI implementation is a good way to proceed.
Understanding Balsam Job Modes
Suppose you need to use Theta to crank through a very large number of
small simulations. The MPI job mode of Balsam works by launching several
concurrent apruns and monitoring the status of each
process. This MPI job mode is the only way to run codes built with
Cray MPI, because initializing communications requires coordinating with
Cray's Application Level Placement Scheduler (ALPS). When applications
use at least 4 nodes and take 1 minute to run, the MPI job mode of
Balsam poses no bottlenecks to your workflow.
For smaller or shorter jobs, we start to feel two limitations of this launch mode. First, Theta supports a maximum of 1000 concurrent apruns per job. That means even if you have 2 million+ single node calculations, each batch job must be limited to 1000 nodes. A natural workaround with Balsam is to submit several 800 node MPI-mode jobs, and allow them process your workload cooperatively.
The second
limitation of the MPI job mode is a 100 ms delay between subsequent
application launches. Assuming a target maximum of 800
apruns sustained in an 800-node job, it will take about 80
seconds to ramp up to full utilization. If applications take several
minutes to run or their completion is fairly staggered, this latency is
hardly noticed. For significantly faster applications, you might find
some that some fraction of the nodes are idle on average, due to
application startup time losses.
Generally speaking, the MPI-mode becomes a bottleneck for very small or
short runs. Balsam's serial job mode can be much faster
and scale to the entire machine in a single allocation, because the
launcher wraps the execution of all tasks under a single MPI runtime
launched at the beginning of the job. Unfortunately, the serial job mode
only works for OpenMP or single-node codes built without Cray MPI, for
the ALPS-startup reason mentioned above. Up til now, if your code used
MPI to scale, you were stuck with the MPI job mode.
Containerizing NWChem 6.8+OpenMPI
If we plan to run single-node instances of our MPI app, we still want
MPI for parallelism across CPU cores. If we could avoid linking the code
with the Cray MPI stack, we would be able to run in Balsam's
serial job mode, since a generic MPI initialization would
be unaware of the interconnect and not try to reach ALPS. Here, I'll
show how Singularity can be leveraged on Theta to run an application
built with generic MPI inside a container.
The example we'll follow here is for NWChem 6.8. Fortunately, Dr. Apra
at PNNL has contributed several examples of Dockerfiles for NWChem
builds to the official Github repository. We'll start with a build that
uses the generic sockets ARMCI
implementation.
This Dockerfile builds NWChem in a Fedora image with completely generic
OpenMPI and Scalapack libraries, so it should be fairly portable and
interconnect-agnostic. One important change for compatibility with the
Singularity runtime, which does not allow setting UID on Theta, is to
remove the USER instruction from the Dockerfile. Instead
of creating an nwchem user and building inside their home
directory in the Docker image, we'll do it the canonical Singularity
way and build our app under /nwchem of the Singularity
image root path.
Here's the modified Dockerfile that I used:
FROM fedora:27
MAINTAINER Edoardo Apra <edoardo.apra@gmail.com>
RUN dnf -y update \
&& dnf -y upgrade \
&& dnf install -y python-devel gcc-gfortran openblas openmpi-devel scalapack-openmpi-devel tcsh openssh-clients which bzip2 patch make perl findutils hostname git \
&& dnf clean all
WORKDIR /nwchem
ENV NWCHEM_TOP="/nwchem/nwchem-6.8.1" \
PATH=$PATH:/nwchem/nwchem-6.8.1/bin/LINUX64:/usr/lib64/openmpi/bin/:/nwchem/nwchem-6.8.1/QA/:PATH \
NWCHEM_TARGET=LINUX64 \
NWCHEM_MODULES="all python" \
PYTHONVERSION=2.7 \
PYTHONHOME="/usr" \
USE_PYTHONCONFIG=Y \
BLASOPT="/usr/lib64/libopenblas.so.0 -lpthread -lrt" \
LAPACK_LIB="/usr/lib64/libopenblas.so.0 -lpthread -lrt" \
BLAS_SIZE=4 \
USE_64TO32=y \
LD_LIBRARY_PATH="$LD_LIBRARY_PATH:/usr/lib64/openmpi/lib" \
SCALAPACK="-L/usr/lib64/openmpi/lib -lscalapack " \
USE_MPI=y \
USE_MPIF=y \
USE_MPIF4=y \
ARMCI_NETWORK=SOCKETS \
NWCHEM_EXECUTABLE=/nwchem/nwchem-6.8.1/bin/LINUX64/nwchem \
NWCHEM_BASIS_LIBRARY=/nwchem/nwchem-6.8.1/src/basis/libraries/ \
NWCHEM_NWPW_LIBRARY=/nwchem/nwchem-6.8.1/src/nwpw/libraryps/ \
FFIELD=amber \
AMBER_1=/nwchem/nwchem-6.8.1/src/data/amber_s/ \
AMBER_2=/nwchem/nwchem-6.8.1/src/data/amber_q/ \
AMBER_3=/nwchem/nwchem-6.8.1/src/data/amber_x/ \
AMBER_4=/nwchem/nwchem-6.8.1/src/data/amber_u/ \
SPCE=/nwchem/nwchem-6.8.1/src/data/solvents/spce.rst \
CHARMM_S=/nwchem/nwchem-6.8.1/src/data/charmm_s/ \
CHARMM_X=/nwchem/nwchem-6.8.1/src/data/charmm_x/ \
OMPI_MCA_btl_vader_single_copy_mechanism=none
RUN cd /nwchem; git clone -b hotfix/release-6-8 https://github.com/nwchemgit/nwchem.git nwchem-6.8.1 \
&& cd nwchem-6.8.1/src \
&& sed -i 's|-march=native||' config/makefile.h \
&& sed -i 's|-mtune=native|-mtune=generic|' config/makefile.h \
&& sed -i 's|-mfpmath=sse||' config/makefile.h \
&& sed -i 's|-msse3||' config/makefile.h \
&& ls -lrt \
&& make nwchem_config && make 64_to_32 \
&& make -j4
ENTRYPOINT ["/bin/bash"]
The container was built with Docker Desktop for Mac OS, using docker build
-t nwchem-681.fedora.sockets .. After building and pushing to Docker Hub,
getting the Singularity image on Theta required only a simple singularity
pull command.
Registering the Balsam ApplicationDefinition
Let's set up a flexible Balsam App and factory function to dispatch
NWChem runs with this container. The following assumes an activated
Balsam DB and that the current directory contains the pulled Singularity
image file nwchem-681.fedora.sockets_latest.sif. The
ApplicationDefinition's executable will do nothing but call
singularity exec. We leave it up to our BalsamJob factory function
to set up the rest of the command line as follows:
import os
from balsam.core.models import BalsamJob, ApplicationDefinition
HERE = os.path.dirname(os.path.abspath(__file__))
IMG = os.path.join(HERE, 'nwchem-681.fedora.sockets_latest.sif')
def nw_job(path, name, workflow='nwtask', nproc=1):
job = BalsamJob(
name=name,
workflow=workflow,
application='nwchem'
num_nodes=1,
ranks_per_node=1,
post_error_handler=True,
)
inp_dir, inp_filename = os.path.split(path)
bind_str = f'-B {inp_dir}:/nwinput:ro' # read-only Singularity bind path
job.args = f'{bind_str} {IMG} mpirun -n {nproc} nwchem /nwinput/{inp_filename}'
job.save()
return job
if __name__ == "__main__":
ApplicationDefinition.objects.get_or_create(
name="nwchem",
envscript=os.path.join(HERE, 'envscript.sh'),
postprocess=os.path.join(HERE, 'post.py'),
executable="singularity exec",
)
The nw_job function accepts a path to any input file and
ensures that it's visible in the container by setting the appropriate
readonly bind path. From Balsam's point of view, it is launching a
strictly serial (1 node, 1 rank, no-MPI) application. Instead, we pass
the nproc parameter to mpirun inside the
container by crafting the commandline arguments on
job.args. This allows the container's OpenMPI to
parallelize NWChem across cores without Balsam even knowing about it. We
can call this nw_job function from anywhere (a login
node, or inside another running job) to programatically dispatch new
NWChem tasks for given input files. By invoking this script directly, we
ensure that the corresponding ApplicationDefinition named
nwchem is created.
Notice that we also associated this App with a postprocessing step and
an envscript for setting up the Application's environment. The
postprocessing step lets us implement a quick error handling/retry step
for jobs that failed due to an intermittent bug with Singularity on
Theta. Sometimes, the getpwuid system call, which is used
to get the current user's UID and home directory, fails when invoked
from the compute nodes. We can catch the error message and tell Balsam
this was not our fault to try the job again. The post.py
should look as follows (with executable permission bit set!)
#!/usr/bin/env python
from balsam.launcher.dag import current_job
if current_job.state == "RUN_ERROR":
stdout = open(current_job.name+'.out').read() # read output from current workdir
if 'unknown userid' in stdout:
current_job.update_state("RESTART_READY", "detected getpwuid error; retrying...")
The envscript sets LD_LIBRARY_PATH, which is propagated inside the
container (no need for the SINGULARITYENV_ prefix), and sets some
additional environment variables in an attempt to mitigate the getpwuid
issue on Theta.
export SINGULARITYENV_HOME=/home/msalim # your home directory here
export SINGULARITYENV_LOGNAME=msalim # your Theta username here
export SINGULARITYENV_GID=100 # the users group id
export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:/usr/lib64/openmpi/lib # path to OpenMPI in the image
Checking single-node performance inside the container
To verify that the ARMCI sockets+OpenMPI build of NWChem was actually
able to utilize the KNL effectively, I checked the strong scaling of a
water hexamer MP2/aug-cc-pvdz energy calculation up to 64 ranks (1 for
each core on the KNL). I used nw_job to add jobs
with nproc between 1 and 64 ranks, with 3 trials per
number of ranks. The results show a satistfactory speedup of the
calculation as nprocs is increased, which shows that
running OpenMPI inside Singularity in the Balsam serial job mode is
actually working:
| # of ranks | Average walltime (sec) |
|---|---|
| 1 | 1127 |
| 2 | 555 |
| 4 | 287 |
| 8 | 157 |
| 16 | 93 |
| 32 | 64 |
| 64 | 50 |
If you are paying attention, the strong scaling efficiency drops from 90% at 8 cores to 35% at 64 cores. This is not surprising given the small system size, completely unoptimized build of NWChem on a MacBook, and no attention paid to I/O or memory settings in the input file. The important point is that using the container's OpenMPI to scale on the KNL does provide a signficant and reproducible speedup, all the way up to 64 ranks. There is certainly room for optimization here. The input file used for this test is provided below.
start h2o_hexamer
geometry units angstrom
O 0.803889 0.381762 -1.685143
H 0.362572 -0.448201 -1.556674
H 1.668734 0.275528 -1.301550
O 0.666169 -0.420958 1.707749
H 0.236843 0.404385 1.523931
H 0.226003 -1.053183 1.153395
O 2.996112 0.001740 0.125207
H 2.356345 -0.159970 0.813642
H 3.662033 -0.660038 0.206711
O -0.847903 -1.777751 -0.469278
H -1.654759 -1.281222 -0.344427
H -1.091666 -2.653858 -0.718356
O -2.898828 0.065636 0.089967
H -3.306527 0.037245 0.940083
H -2.312757 0.817025 0.097526
O -0.655160 1.814997 0.176741
H -0.134384 1.449649 -0.543456
H -0.526672 2.749233 0.167243
end
basis
* library aug-cc-pvdz
end
task mp2 energy
Acheiving throughput on 2048 nodes
Finally, you should uncomment export
MPICH_GNI_FORK_MODE=FULLCOPY in your Balsam job template
in ~/.balsam/job-templates/theta.cobaltscheduler.tmpl.
This flag mitigates an issue in the Cray MPI stack that arises when
Balsam (or any other application) spawns child processes at scale. You
can now populate your Balsam database with up to millions of NWChem
tasks, and use balsam submit-launch --job-mode=serial to
submit several (up to 20) default queue jobs with no limit on the number
of requested nodes.
I tested the throughput of this setup by populating the Balsam database with 32k identical NWChem jobs. The input deck was for a simple water molecule MP2/aug-cc-pvdz gradient calculation; a single instance of this calculation takes about 9 seconds for the container running on the KNL. A 2048 node job was able to complete 22,765 calculations in 8 minutes without any faults in the workflow. We can use Balsam to extract the job history metadata and get a quick look at the throughput of jobs. To get a first look at throughput, the following code snippet can be used to trace the number of completed job events over time:
from matplotlib import pyplot as plt
from balsam.core import models
timestamps, num_done = models.throughput_report()
plt.step(timestamps, num_done, where="post")
plt.show()
The following bare-bones graph, missing axis labels and all, is obtained from the snippet above:
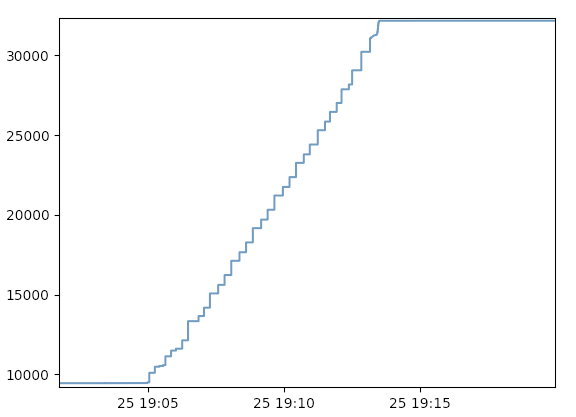
Dividing the 16,384 node-minutes by 22,765 completed tasks, the total
node-time per calculation comes to 43 node-seconds. Given that the
actual walltime spent in NWChem is 9 seconds, there is a substantial
overhead here. The loss in efficiency can partially be attributed to the
FULLCOPY fork mode, which has a significant impact on
subprocess startup time. There is an open ticket with Cray to look at
resolving this issue. We will continue to look at other potential
bottlenecks in the Balsam serial job executor and Singularity startup
time to improve short-task throughput. For the time being and more
realistic problem sizes, this is a promising and already effective
option for running large numbers of single-node MPI calculations at
scale with Balsam.